一种通过水和二氧化碳的共电解产生合成气并具有可调的H2/CO电解产率的全氧化物电解电池
1.研究背景
为了应对与全球温室气体排放相关的气候变化和可持续性挑战,我们非常希望采用经济上可行和高效的技术来减少二氧化碳排放或/并将二氧化碳转化为高价值化学品。高温固体氧化物电解电池(SOECs)提供了一个特别有趣的解决方案。如果由可再生电力供电,SOECs可以用于二氧化碳和水的共电解,以产生“绿色”合成气,然后可以用作后续生产各种绿色化学品和燃料]的原料。特别是,SOECs产生的绿色合成气可以直接送入费托托(F-T)反应器,合成许多目标化学物质,包括甲醇、醛、碳氢化合物。
重要的是,在F-T过程中,合成气中的H2/CO比值对目标产物的产率和合成气的转化过程起着重要的作用。例如,H2/CO比为2的合成气是甲醇合成的最佳合成,而H2/CO比为1有利于醛的生产。理想情况下,还应建立对SOEC共电解输出的H2/CO比值的可调控制,以使产物合成气组成与后续下游合成气转化过程的目标要求相匹配。一般来说,固体氧化物共电解过程中产生的出口气体组成取决于阴极的催化性能、入口H2O/CO2比和工作温度,以及施加的电流密度/电压。近年来,人们对对H2O/CO2还原催化活性高的阴极材料进行了广泛的研究。例如,最先进的Ni-YSZ陶瓷阴极,具有高电催化活性、低制造成本和地球丰富的前驱体,是一种很有前途的传统YSZ的电极材料。然而,在现实的情况下,镍基阴极在高电流密度下容易被氧化,导致裂解并最终失效的。此外,镍陶瓷基电池通常需要H2和/或CO注入阴极除了水和二氧化碳避免团聚或氧化的镍粒子在高温下,尽管这可以降低整体效率,增加系统的复杂性,并限制H2:CO比率的范围可以获得的电池。将一些高活性的过渡金属元素,如Fe、Co等,引入镍陶瓷阴极,以提高CO对H2O-CO2共电解的选择性。然而,CO产量的增加主要是由于反向水气位移反应(RWGS)的加速。钙钛矿结构氧化物作为镍陶瓷阴极的替代品,由于其优越的氧化还原稳定性和抗结焦,已成为很有前途的阴极材料。因此,它们可以在水和/或二氧化碳环境中直接运行,而没有安全气体(H2和/或CO),并且通常显示出增强的抗结焦能力,这对于高电流密度操作或当需要生产富含CO的合成气组合物时尤为重要。钙钛矿基SOEC阴极一般基于LaCrO3、LaFeO3、SrTiO3和Sr2Fe1.5Mo0.5O6体系。然而,从这些阴极中获得的电化学性能还需要进一步的改进。与传统的Ni-YSZ陶瓷阴极相比,“全氧化物”钙钛矿阴极普遍较低的电导率和水和/或二氧化碳分裂的催化活性降低,受到特别的挑战。
虽然阴极组成和结构对输出H2/CO比的影响一直是一些研究的主题,但较少关注操作条件的影响,包括操作温度、施加的电压/电流和原料气中的H2O/CO2比。在这里,我们建立了如何使用这些关键的操作变量来对全氧化物基共电解电池中的输出H2/CO比值进行可调谐的控制。虽然在大多数基于SOEC的共电解细胞中获得富H2的合成气组成(例如H2/CO > 2)没有问题。我们基于La0.7Sr0.3Fe0.9Ni0.1O3的钙钛矿氧化物阴极,即使在高度富含CO的生产条件(H2/CO = 0.1)下,也能实现稳定的电解操作,这表明这种电池结构可能是未来共电解应用的一个有前途和灵活的候选者。
2.成果简介
在固体氧化物电解电池(SOEC)中实现了可调的H2/CO和无焦操作的高速率生产合成气。在操作前,使用控制预还原LSFNi La0.7Sr0.3Fe0.9Ni0.1O3-δ(LSFNi)阴极触发平均尺寸镍铁合金纳米颗粒~45 nm均匀分布在LSFNi主干上,使水和二氧化碳有效地共电解生成H2和CO。在1.5 V,750℃达到~1.0A cm−2, 850℃达到~2.4A cm−2,接近100%法拉第效率。我们证明了通过操纵原料气的H2O/CO2比值、工作温度和电流密度,证明了将输出的H2/CO比值调整近两个数量级(从~0.1到~7)的可行性。>100 h的稳定运行,没有碳沉积的证据,但高电流密度运行导致阳极/电解质界面明显恶化。
3.图文导读

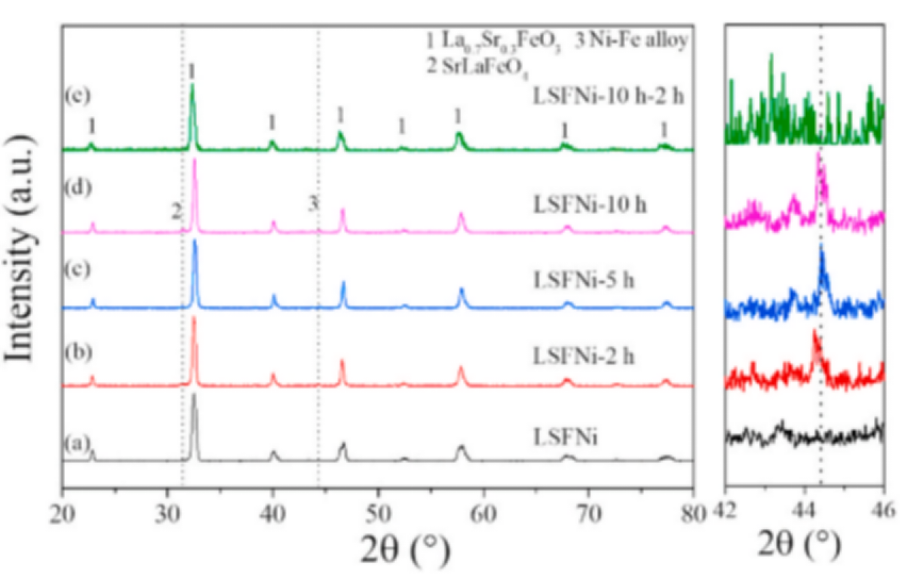
图1 煅烧的LSFNi粉体(a).的XRD模式LSFNi在850◦C和5% H2-Ar条件下进行不同时间还原后的XRD图(b-d)。LSFNi样品在还原10 h后,在850℃的空气中再氧化的XRD图(e)。右侧为42°到46°之间的放大结果。

图2 (左图)LSFNi粉末在850◦C下还原不同时间后的扫描电镜。(A)0h(原始粉末)
(B)2h(C)5h(D)10h(E)还原处理5h后在850◦C空气中再氧化2h。(右图)LSFNi在(A、C、E)前和(B、D、F)还原5h后的扫描电镜和TEM。
在1100℃的空气中煅烧10 h后,得到纯具有菱形对称的LSFNi钙钛矿粉末(编号167),如图1 (a).所示众所周知,一些过渡金属元素,如Fe、Ni、Co,在适当的还原气氛下,可以很容易地从一些钙钛矿体系中溶出。为了探索LSFNi体系中的溶解过程,将LSFNi粉末分别在850℃的5% H2-Ar中还原2h、5h和10 h。图1(b-d)显示了经过这些还原处理后的LSFNi材料的XRD模式。所有LSFNi样品均保留La0.7Sr0.3FeO3-δ钙钛矿主相,并加入少量SrLaFeO4和镍铁合金二次相,表明LSFNi氧化物具有良好的相稳定性。在5%H2-Ar条件下,镍铁合金信号(PDF: #88-1715)在44.3°左右的XRD强度随着暴露时间的增加而增加,表明LSFNi表面有较高比例的溶解纳米颗粒。此外,在850℃的空气中再氧化2小时后,二次相可以重新加入到钙钛矿骨架中(图1(e)),这表明LSFNi具有强大的氧化还原稳定性。图2显示了原始LSFNi粉末分别在850℃下还原2h、5h和10 h后的形态及其随后的形态演变。经5%H2-Ar处理后,LSFNi粉末的表面形貌变化明显。与原始(未还原)粉末的清洁表面形貌相比,还原的LSFNi粉末上纳米颗粒的分布相对均匀,并且随着处理时间的增加,纳米颗粒的数量逐渐增加。在850℃的空气中再氧化2小时后,大部分纳米颗粒重新结合到母氧化物相中,表明在LSFNi晶格中过渡金属阳离子扩散容易且可逆。然而,与新鲜的LSFNi相比,再氧化的LSFNi样品的表面非常粗糙,这表明在溶解过程中,微观结构仍存在一些不可逆的变化。
致密的LSFNi条也处理在850℃,5%H2-Ar下处理5h研究纳米颗粒沉淀的形成和大小分布,平均~45纳米直径(如左图2(B)所示),并证实了固态动力学的溶解过程是快速的,即使在一个相对密集的紧凑。图左2(A)显示了新鲜的LSFNi氧化物的一个相对光滑的表面,尽管观察到一些属于金纳米颗粒的较小的颗粒。此外,我们得到了原始LSFNi晶格的(101)平面间距为0.394 nm,如右图(E)所示。相比之下,LSFNi衬底(点2)中的主要母相的Ni/Fe比比新鲜粉末(9.7比16.4)大大降低,溶解的纳米颗粒(点3)中的Ni/Fe比估计为1.6。这些结果支持了富镍铁合金纳米颗粒从LSFNi主链中溶解的结论。
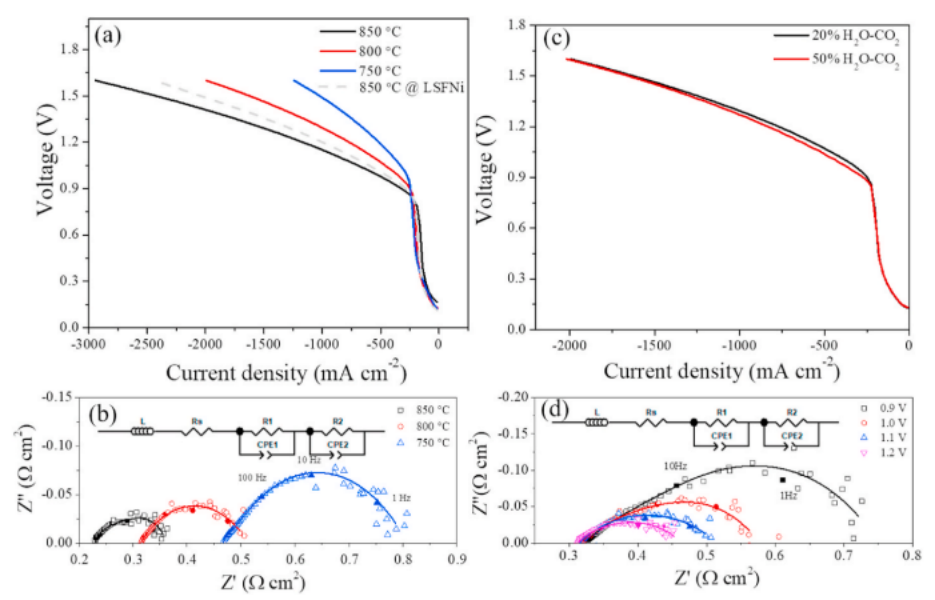
图3 LSFNi-GDC/LSGM/PBCC-GDC细胞对H2O-CO2共电解的电化学性能(a);在750-850℃下共电解1.1V下的I-V曲线和奈奎斯特图(b);不同蒸汽含量的H2O-CO2共电解在800◦C下的I-V曲线(c);不同应用电位下20 vol% H2O-CO2在800◦C下的EIS曲线(d)。
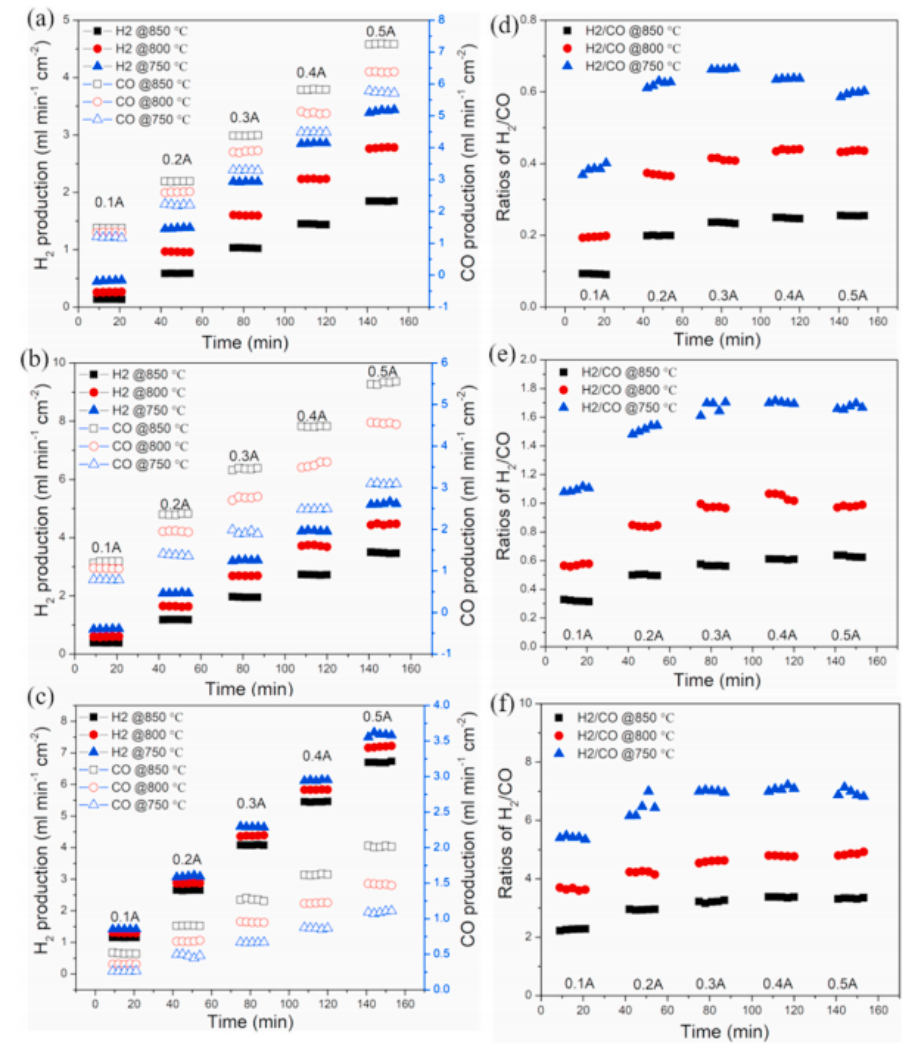
图4 不同条件下H2O/CO2共电解的实验数据。(a、d)为10%的volH2O-CO2;(b,e)20%volH2O-CO2;(c,f)50%volCO2。

图5 在20%体积下测量两种不同电流密度下共电解电池的长期稳定性。H2O-CO2和800℃。(a)−500 mA cm−2;(b)−1000 mA cm−2。
在LSFNi-GDC在5% H2-Ar中处理之前,还在850℃和20% H2O-CO2条件下进行了电解细胞的性能,结果如图3 (a).所示虽然在850◦C下获得了−2450 mA cm−2的高电流密度值,但该值仍低于镍铁合金修饰的LSFNi阴极。由于镍铁合金纳米颗粒具有较高的催化活性,因此具有优异的电池性能。EIS数据使用Zview软件进行分析,使用LRS(R1- CPE1)(R2-CPE2)等效电路,如图3(b)插图所示。在1.1 V的应用电位下,在850、800和750℃下进行共电解操作时,我们分别获得了0.14、0.21和0.35 Ω cm2的Rp值(图3(b))。水通过对称电池测试得到的PBBC-GDC阳极的Rp值相比,这些结果表明,大部分的全电池的极化电阻可以归因于阴极电极。Rp随温度的升高而降低,这与催化和/或电化学活化相一致。增加蒸汽含量也能提高性能,如图3(c)所示,这与水还原相对于二氧化碳还原的一般更快的动力学相一致。
图4总结了在我们的SOEC中H2O-CO2共电解中实际实验测量的H2和CO产物气体组成(通过在线GC获得)。图中的比较。实验结果与热力学预测结果定性一致。如图4(a、b、c)所示,H2和CO的产量随电流的增加呈线性增加。此外,随着工作温度从750℃增加到850℃,水向H2的转化降低,而CO2向CO的转化增加。与热力学预测一致,产物H2/ CO比值随温度的升高而显著降低,如图4(d,e,f)所示,而电流的影响很小。重要的是,温度对实验测量的H2/CO比值的影响似乎比热力学预测的影响要大得多,这表明动力学也很重要。而热力学分析预测,实验结果表明,在相同的范围内(从~0.1到~7),H2/CO比值发生了近70倍的变化。有趣的是,在所有情况下,实验测量的H2/CO比值都远远高于热力学预测(除了10%vol。在850◦C下的H2O-CO2,实验结果接近热力学极限)。我们将此归因于水电还原相对于二氧化碳电还原更快的快速反应动力学,以及RWGS反应的有限动力学。电解产物在阴极电极内的停留时间可能不足以使完全的RWGS反应平衡,因此产物流H2组成相对于热力学预测而升高。进一步研究了H2O-CO2共电解电池在低、高电流密度操作下的长期性能,评价了Ni-Fe改性LSFNi阴极的稳定性。如图5所示,低电流密度操作与高电流密度操作的电压衰减率显著降低(即−500 mA cm−2的0.13mVh−1与−1000 mA cm−2的0.72mVh−1)。在较高的电流密度下,SOECs的降解通常会增加,这通常归因于两个主要的潜在原因。一个原因是由于快速析氧导致阳极/电解质界面恶化,而另一个原因是阴极可能结焦。
4.总结与展望
综上所述,通过控制还原原位解出可以制备镍铁纳米颗粒修饰的LSFNi阴极,在H2O-CO2共电解操作下具有优异的性能。细胞可用于调整H2/CO产品比例在大范围(从~0.1~7)同时保持良好的稳定性和无焦炭操作,尽管一些退化观察高电流密度(−1000 mA cm−2)由于孔隙形成电解质/阳极界面。将电化学反应与RWGS平衡耦合的热力学考虑定性地解释了共电解操作下H2/CO产生的趋势,尽管动力学因素也很重要。由于水电还原比二氧化碳电还原要容易得多,实验测量的H2/CO比值总是高于热力学预测,除了在高温和低PH2O的极端情况下,实验结果接近热力学极限。在这里的所有案例研究中,总体共电解法拉第效率被测量为~100%(在实验误差范围内),合成气产生速率随预期的施加电流线性增加。结果表明,LSFNi阴极和LSFNi-GDC/LSGM/PBCC-GDC细胞结构作为一种有前途的SOEC系统,从二氧化碳和水共电解产生稳定、可调、高速率的“绿色”合成气。
5.文献信息
An all-oxide electrolysis cells for syngas production with tunable H2/CO yield via co-electrolysis of H2O and CO2. (Journal of Power Sources. 2021). https://doi.org/10.1016/j.jpowsour.2020.228887
6.作者简介
卞刘振(1990.04),副教授,硕士生导师男,汉族,河南周口人,内蒙古科技大学材料与冶金学院专任教师,主要研究新能源材料与器件方向。